
| Table of Contents |
|---|
Information on data standards implementation
|
Considerations for data standardisation
How to Move Forward with a Standardisation Effort Creating new standards and new conventions are by no means easy tasks. However there are some tricks that should be considered when looking into standardising a new study type or new endpoints within studies where some standards already apply. As your organisation becomes SEND-aware and develops its go-forward strategy, it must be acknowledged that SEND is being used for both operational as well as submission purposes. Understanding how your organisation intends to respond to the standard is important. Perhaps you will use it only operationally or only to participate in pilot activity until it becomes a requirement. Or, you may intend to submit one package for one study in your next update. Alternatively, you may plan to submit a package for the majority of the studies in your next NDA or IND. For a vendor or a CRO, the need is to be as ready as your first client request.
|
| Introduction |
|---|
| Advances in informatics and data standards are helping to optimise nonclinical data utility to meet demands for more effective and efficient drug development. The CDISC Standard for Exchange of Nonclinical Data (SEND) team is currently working to standardise nonclinical study types. The FDA, as well, is establishing regulatory guidance for the electronic submission of these nonclinical studies. Collectively, these efforts make this a formative time for stakeholders of these standards. As a result, the FDA’s Center for Drug Evaluation and Research (CDER) in conjunction with representatives across the industry have formed a collaboration aimed at addressing common issues with regards to nonclinical data management, data standards, and standards implementation. A Nonclinical Working Group within the collaboration identified several areas of interest and formed teams to work on each one. A summary of the Nonclinical Working Group and its various work-group teams is provided in the resource list at the end of this document. One of the working groups, the Nonclinical Standards Roadmap Team (Roadmap Team), identified a need to orient newcomers to standards, set priorities for nonclinical data, and consider ways to facilitate future standardisation efforts. Similar to what has already been done for clinical data by CDISC, the group realised a need to develop a strategy on how to prioritise, maximise, and revamp the standardisation efforts for nonclinical data. The team aimed to create a nonclinical standards roadmap which could be useful to all, whether one is a novice or working on future steps in nonclinical data standardisation. The Roadmap Team focused on the following objectives: • Identify points to consider for implementing a standard with a goal to minimize redundancy and maximise collaboration • Highlight important steps of the data standardisation process • Prioritise the nonclinical data types • Design a survey and obtain results from members of industry to further understand the priorities of the many stakeholders of nonclinical data • Compile list of key standards resources (Links for Stakeholders below) for implementing the best approach for future standardisation efforts • Inform stakeholders about the types of standards and standards organisations that are available and the potential role that stakeholders can play in standards development |
| Considerations for Implementing Standards |
|---|
Standards implementation poses many challenges for organizations of all sizes. The PHUSE Nonclinical Working Group prioritized it as a major challenge and formed an Implementation Team devoted to addressing questions about standards utilization. Their work culminated in a Frequently Asked Questions (FAQ) resource which is posted at the following link (SEND Implementation FAQ). The Roadmap Team considered that many organisations are in early stages of implementation, and therefore also documented questions to consider during the process.
So, what are the necessary steps to become compliant with requirements for electronic data? Where should you begin? This may seem obvious, but it cannot be repeated too often: Start with what you know, and accept that the first iteration of your standardization exercise will not be 100%. If it takes you to 80%, at least you are only 20% from your target instead of where you started.
This approach would require a moderate analysis, and result in an extensive implementation impact. For example, many LIMS systems would require some degree of customization for this approach to work.
This approach would require an extensive analysis, and result in a moderate implementation impact. For example, it may be possible to enforce the use of controlled terminology, but not the final output format of the electronic data.
This approach would require a moderate analysis, and carries with it the potential for extensive implementation impact. For example, if the data standard is only enforced at the end when creating the submission data, the collected data face a conversion process that likely ends up being a manual exercise requiring specialist knowledge. A single approach may suffice. However, once your analysis is complete, a combination of the above approaches may be required. Once the approach(es) is identified, the following must be considered and/or planned for:
While there is a lot that must be considered, planned, and accomplished to implement a standard, there are extensive benefits to data standardisation and overall increases in the quality of the data. Data discernment, reproducibility, exchange, analysis, cross-study analysis, storage, etc., will significantly improve as more standards are developed and implemented across the industry. |
| Setting the right foundation for communication |
|---|
SME engagement is essential to the success of the standards implementation process. In order to capture your audience, you must address them at their level of awareness. Below you can find some questions and answers that can help you when delivering the standardization message to the subject matter experts (SME) in your organization. What does your audience know about SEND? SEND, the Standard for Exchange of Nonclinical Data, is an implementation of the CDISC Standard Data Tabulation Model (SDTM) which specifies a way to present nonclinical data in a consistent format. 2. What does your audience know about PDUFA V and the draft guidance that will allow the FDA to make requiring SEND output a regulation? 4. What does your audience know about FDA projects like the Janus Data Warehouse? (Stakeholders details below) |
| Future Standardisation |
|---|
How to Move Forward with a Standardisation Effort Creating new standards and new conventions are by no means easy tasks. However there are some tricks that should be considered when looking into standardising a new study type or new endpoints within studies where some standards already apply. As your organisation becomes SEND-aware and develops its go-forward strategy, it must be acknowledged that SEND is being used for both operational as well as submission purposes. Understanding how your organisation intends to respond to the standard is important. Perhaps you will use it only operationally or only to participate in pilot activity until it becomes a requirement. Or, you may intend to submit one package for one study in your next update. Alternatively, you may plan to submit a package for the majority of the studies in your next NDA or IND. For a vendor or a CRO, the need is to be as ready as your first client request. |
| Prioritisation of Nonclinical Data |
|---|
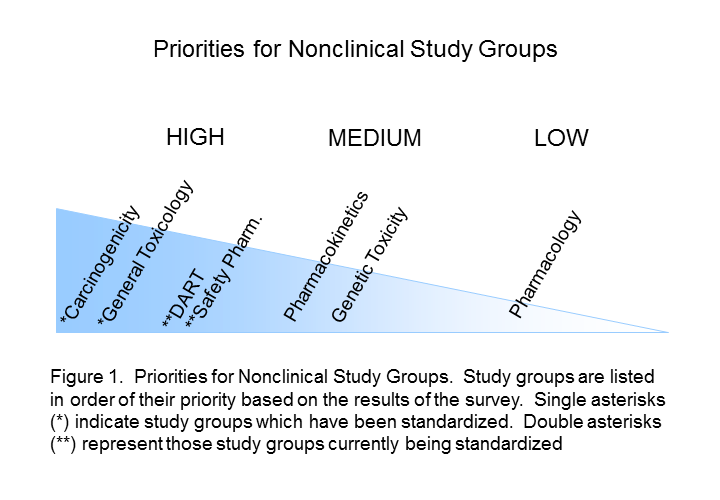
The Standards Roadmap Team created a survey for members of industry to understand the priorities of the many stakeholders of nonclinical data. We asked them to consider the entire expanse of nonclinical studies listed for Module 4 of the eCTD. Responders rated the study types with a scale of high, medium, or low priority or had the option of leaving it blank (indicative of no preference). A spot to comment on rationale for the preference was provided. Finally, responders were asked to indicate whether they were responding as an individual or on behalf of their organization. Results Respondents consisted of both companies/institutions (n=4) and individuals (n=11) throughout industry. Overall, there was no significant difference between company and individual respondents. The results of the survey indicated that Carcinogenicity and General Toxicology were the highest priority study groups (Figure 1). This was not surprising and supports previous decisions by the SEND team to standardise these first. Developmental and Reproductive Toxicology (DART) and Safety Pharmacology were rated the next highest priorities. Indeed, standards for these two study groups are currently being developed. Pharmacokinetics studies were considered to be a medium priority, along with Genetic Toxicology studies. Respondents differed on the utility of pharmacology studies, which received both high and low priority status, hence it is labeled as lower priority in Figure 1.
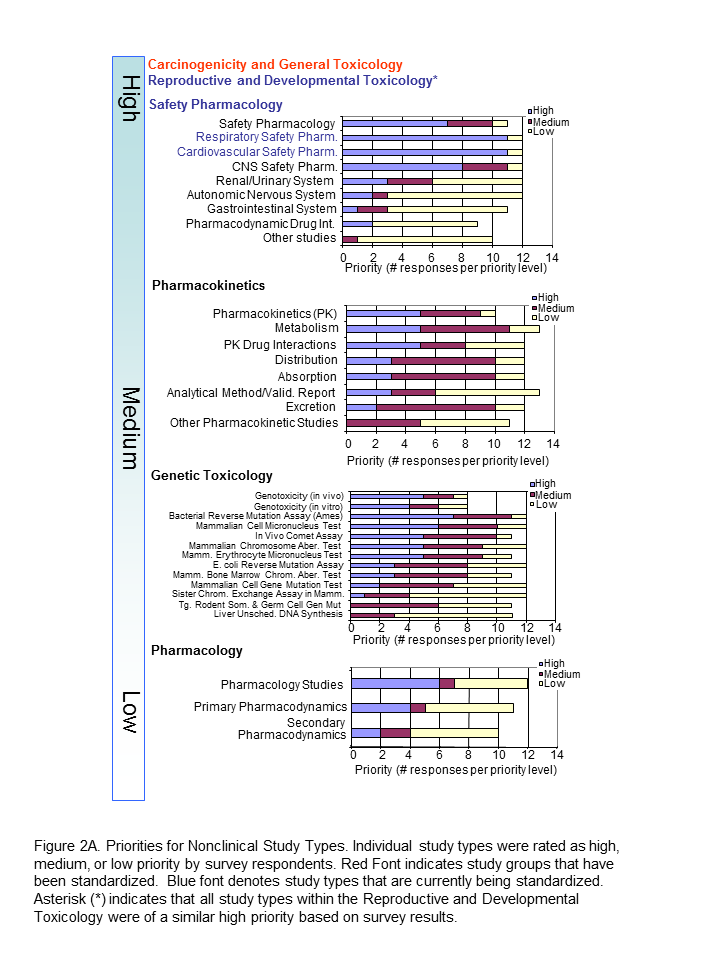
The Roadmap team recognised value in keeping study types grouped since conventions of standardization may be applied to several study types within a study grouping. Study types for developmental and reproductive toxicity (DART studies: Segment I, II, III) were similar in their perceived high priority. Safety Pharmacology had both high and low priorities among their study types (Figure 2A). Note that Respiratory and Cardiovascular Safety Pharmacology study types received the highest priority and are presently being standardized. CNS Safety Pharmacology also rank high, while Renal, Autonomic Nervous System and Gastro-Intestinal systems are lower. Pharmacokinetic and Genetic Toxicity study groups received high/medium scores across their respective study types.
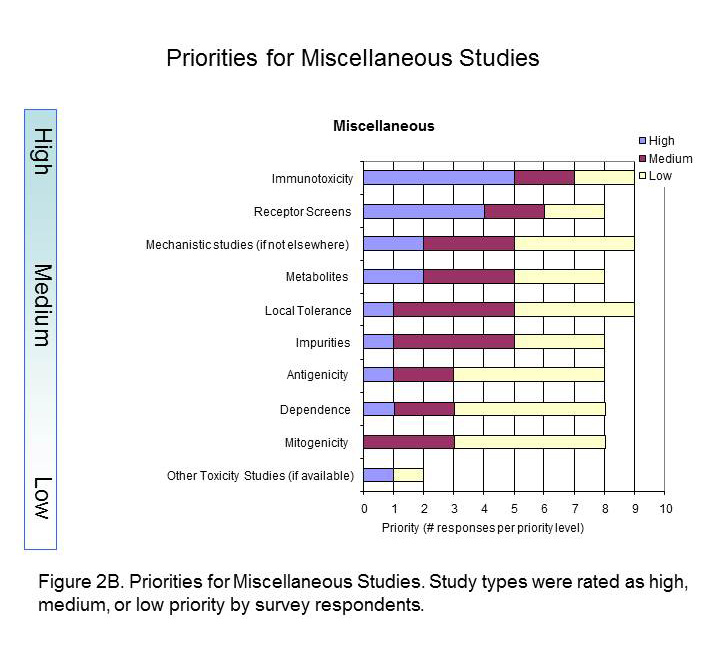
Figure 2B captures prioritisation of miscellaneous studies. Immunotoxicity and Receptor Screens were ranked highest, while all other studies take medium to lower importance.
A prioritisation of several data elements which can be applied across study types is also provided in Figure 3. Respondents considered creating standards for Historical Control data, Study Design/Logistics, and Clinical Signs of highest importance. Formulation Data, Biomarker Incorporation, and Metadata content also were important for standard implementation.
Survey Conclusion |

| Summary |
|---|
| Future Directions Large volumes of nonclinical data are being produced to provide evidence of the safety of investigational drugs. Currently, these data are recorded, compiled, and transferred in various forms. This is not an impediment to data access as long as our disparate systems allow the visualization to be electronic and standardized. By taking the many and varied processes, systems, lexicons, and formats used in the collection and reporting of drug development data across our industry and collaborating to create a common standard for visualizing it, we will improve our access to data. By basing both clinical and nonclinical data on the same data tabulation model, the science of predictive toxicology can be furthered, proving hypotheses and moving theory to reality. Flexible standards for collecting, conveying, and submitting data provides access to a vast amount of information. The Roadmap Standards team is now undertaking an exercise to identify the possibilities that exist with such a model. Ultimately, the goal is to improve the collection and transmission of nonclinical information and to consider existing models which may give us access to more data. The benefits and challenges to such model are being considered by many stakeholders as they plan to test such a model in a pilot. Conclusion The nonclinical scientific community has been harmonizing data collection methods and data analysis for many years. Some of these efforts have resulted in powerful research and predictive tools and others in publications of harmonized nomenclature and diagnostic criteria for nonclinical lesions. These efforts should be leveraged when looking into developing a standard. The challenge can be that some of these harmonized ontologies (‘scientific synonym dictionaries’) are considered proprietary and therefore not easily accessible. Ideally the companies owning these can find an appropriate business incentive to participate in a standardization effort, such as becoming a recognized standards training and implementation partner for the industry or as a valued software vendor. A harmonization effort is the first step on the way to developing a new standard. One can quite easily get lost in translation, so to speak. Many people believe that once you have harmonized the nomenclature, it is a standard, but it takes quite a bit more. A standard should include both uniquely defined concepts that ideally can be applied throughout the nonclinical (and potentially the clinical) world as well as recommended or preferred terminology for every concept. Whether concepts or terminology come first is the ‘chicken and egg’ discussion. In truth, concepts can help define terminology and vice versa. A standard also requires adoption, use, maintenance, and support. The nonclinical scientific community should strive to have broadly adopted standards to enable a common understanding of the science. In the end, everyone is dependent on others understanding our data. This, however, requires up front “buy in” from multiple stakeholders, and there should be incentive to adopt the standard such as a regulatory push, an unmet scientific need, a measurable saving in time to reporting, man hours per study, etc. A standard must evolve according to science and business needs. This necessitates ownership to its continuous maintenance and support as can be provided by a Standard Development Organization (SDO). Various SDOs exist as organizations or industrial consortia, build on expertise and interest from the people involved, with an established infrastructure allowing informed decision-making about the development of a standard. A standard can exist on many levels. You may have your own personal standard for how you do things or a department or company could have a standard. Incentive may help the tricky task of advancing a standard to the next level of adoption. Why should a company adopt a global standard or why should you adopt a colleague’s standard for that matter? It is about convincing the right people, in the right way, that having well defined standards increases the effectiveness, scientific quality and, ultimately, the regulatory compliance of your work. The Roadmap Team believes that improved access to data through standards provides benefits on many levels. |
Links for Stakeholders |
|---|
SEND Implementation guide CDISC terminology: NCI Thesaurus
Study Data Standards for Submission to FDA webpage:
Draft guidance: Providing Regulatory Submissions in Electronic Format - Standardised Study Data Study Data Specifications v2.0 Prescription Drug User Fee Act (PDUFA) V
SEND Implementation User Group team (PHUSE Nonclinical Working Group) |